




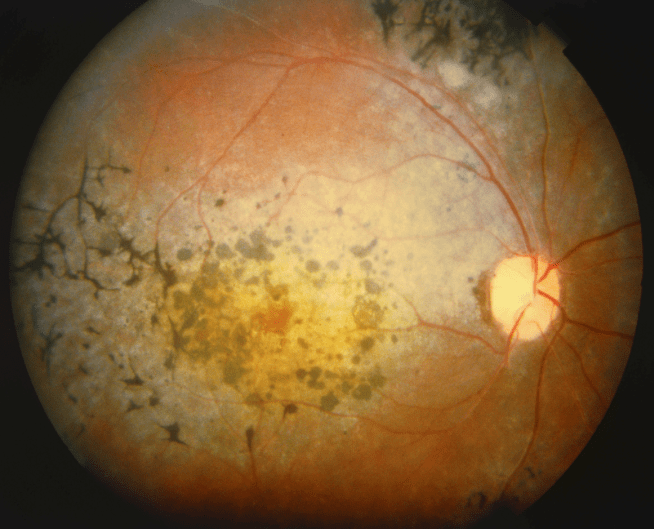
La retinosis pigmentaria (RP) es la distrofia retiniana más frecuente, caracterizada por ser una patología degenerativa, bilateral, hereditaria y causante de una disminución paulatina de la agudeza visual que puede llegar a ser severa e incapacitante. Su prevalencia es de 1/3000 a 1/5000 habitantes.
¿Qué causa la enfermedad Retinosis Pigmentarla (RP)?
La transmisión de unas generaciones a otras sigue diferentes patrones de herencia, si bien los casos esporádicos son los más frecuentes:
- Herencia autosómica dominante: Normalmente se da en todas las generaciones de una familia. Cada persona afectada tiene un progenitor afectado y un 50% de posibilidades con cada hijo de que éste herede el alelo mutado y desarrolle la enfermedad autosómica dominante.
- Herencia autosómica recesiva: Normalmente no se da en todas las generaciones de una familia. Cada persona afectada tiene generalmente ambos progenitores sanos pero portadores del alelo mutado. En los hijos de una pareja en la que ambos son portadores puede ocurrir:
—-50% de probabilidades de ser portadores de una copia del alelo alterado (no expresan enfermedad pero pueden transmitirla a su descendencia)
—-25% de probabilidades de tener dos copias del alelo alterado y desarrollar enfermedad autosómica recesiva
—-25 % de probabilidades de heredar dos copias del alelo normal y no desarrollar la enfermedad ni ser portador
- Herencia recesiva ligada al sexo(cromosoma X): Sólo padecen la enfermedad los hombres. Las probabilidades de tener un hijo afecto para una madre portadora son del 50 % y sus hijas tienen también el 50 % de probabilidades de ser portadoras.
En la RP, se han identificado aproximadamente 12 genes responsables de a RP tipo dominante, unos 6 genes responsables de la RP tipo recesivo, y 6 relacionados con la herencia ligada al sexo.
El consejo genético es fundamental, para saber si existe riesgo de que en una familia pueda repetirse la enfermedad, o bien aparecer por primera vez alguna enfermedad hereditaria.
¿Qué es la Retinosis Pigmentaría (RP)?
Clínicamente, el rasgo característico es la pérdida progresiva de fotorreceptores y otras células retinianas por apoptosis (muerte celular), lo que conlleva una alteración en la funcionalidad de la retina, que por lo general se hace evidente cuando la enfermedad se encuentra ya en una fase muy avanzada.
Los fotorreceptores principalmente afectados son los bastones, fundamentales en la visión nocturna, con la subsiguiente afectación de los conos, que son los fotorreceptores que nos permiten ver en condiciones de luminosidad e identificar los colores.
La edad de aparición es muy variable, siendo los síntomas más característicos:
Nictalopia (mala visión nocturna, dificultad para adaptación a oscuridad o cambios luz-oscuridad), fotofobia (intolerancia anormal a la luz), disminución progresiva del campo visual que en estadíos avanzados conlleva visión en túnel.
Como hemos explicado antes, la causa es genética. Pero hay factores ambientales que pueden influir directamente sobre el gen. De tal manera que la evolución es muy distinta de unos pacientes a otros, incluso perteneciendo a la misma familia y teniendo la misma mutación genética conocida. Por lo tanto, predecir la evolución de la enfermedad es muy difícil.
¿Qué tratamiento tiene la RP?
El tratamiento se basa en enlentecer el proceso degenerativo protegiendo la retina de la luz solar, la administración de complejos vitamínicos y tratando las complicaciones (cataratas y edema macular). Ayudar a los pacientes a hacer frente a las consecuencias sociales y psicológicas de la ceguera es una parte fundamental del tratamiento.
A día de hoy, se apuesta por la investigación de nuevas estrategias terapéuticas, como la terapia génica, basada en introducir material genético en las células para compensar genes anormales o para producir una proteína beneficiosa.
Se está desarrollando un estudio clínico multicéntrico europeo, que valora la eficacia del chip retiniano IRISRII. Es un implante retiniano reversible, que capta las imágenes proyectadas a la retina a través de una minicámara instalada en unas gafas que forman parte del sistema.
La RP puede aparecer como una enfermedad aislada, o también en el cuadro de una asociación sistémica, como el Síndrome de Usher, trastorno angustiante que combina ceguera con sordera profunda en niños.